Manual
PySNAIL provides an application programming interface (API) in Python for developers or bioinformaticians who wants to control more parameters used in the analysis.
Correct Expression
import os
from pysnail import Dataset, qsmooth
xprs = os.path.realpath('sample_data/expression.tsv')
groups = os.path.realpath('sample_data/groups.tsv')
dataset = Dataset(xprs, groups, **{'index_col': 0, 'sep': '\t'})
xprs_norm, qstat = qsmooth(dataset, aggregation='auto', threshold=0.2, cutoff=0.15)
xprs_norm.to_csv(os.path.realpath('pysnail_out.tsv'), sep='\t')
Information of Input Data
print(dataset)
Statistics of Qsmooth
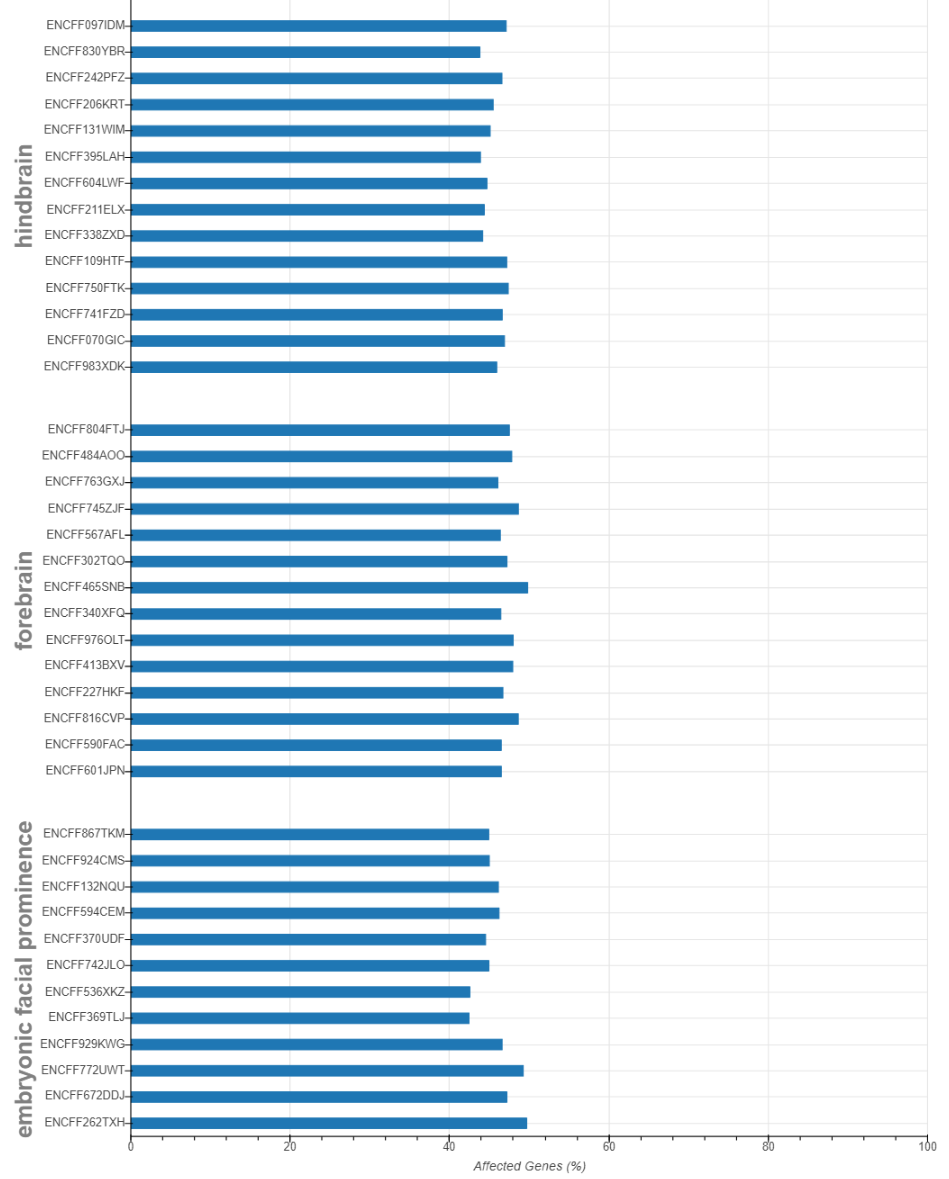
Get number of affected genes for each samples.
qstat.num_affected_genes
Result:
Group Sample
Embryonic Facial Prominence ENCFF132NQU 0
ENCFF262TXH 0
ENCFF369TLJ 0
ENCFF370UDF 0
ENCFF536XKZ 0
...
Stomach ENCFF052DOQ 0
ENCFF288JNN 0
ENCFF355MOU 0
ENCFF691EQW 0
ENCFF972NMO 2730
Length: 126, dtype: int64
Get number of affected samples in the dataset.
qstat.num_affected_samples
Result:
1
Get affected genes for each sample.
print(qstat.num_affected_samples)
Result:
Group Embryonic Facial Prominence \
Sample ENCFF132NQU ENCFF262TXH ENCFF369TLJ
ENSMUSG00000082905 False False False
ENSMUSG00000026174 False False False
ENSMUSG00000031293 False False False
ENSMUSG00000062458 False False False
ENSMUSG00000083793 False False False
... ... ... ...
ENSMUSG00000015093 False False False
ENSMUSG00000098607 False False False
ENSMUSG00000102632 False False False
ENSMUSG00000093969 False False False
ENSMUSG00000050876 False False False
Group \
Sample ENCFF370UDF ENCFF536XKZ ENCFF594CEM ENCFF672DDJ
ENSMUSG00000082905 False False False False
ENSMUSG00000026174 False False False False
ENSMUSG00000031293 False False False False
ENSMUSG00000062458 False False False False
ENSMUSG00000083793 False False False False
... ... ... ... ...
ENSMUSG00000015093 False False False False
ENSMUSG00000098607 False False False False
ENSMUSG00000102632 False False False False
ENSMUSG00000102632 False False False False
ENSMUSG00000093969 False False False False
ENSMUSG00000050876 False False False False
Diagnosis of Qsmooth
Make bar plot on number of affected genes for each sample
from pysnail import bokeh_affected_barplot
bokeh_affected_barplot(dataset, qstat, 'output')
Result (the result shown here is excerpted from the analysis in the manuscript instead of from the sample data. ):